Interpolative Approach
We start with writing down the Anderson impurity Hamiltonian
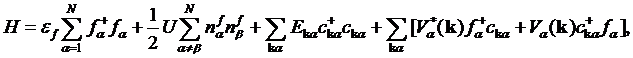 (1)
(1)
describing the
interaction of the impurity level ![]() with
bands of conduction electrons
with
bands of conduction electrons ![]() via hybridization
via hybridization ![]() .
. ![]() is the Coulomb repulsion between different orbitals in the
is the Coulomb repulsion between different orbitals in the ![]() –band. In
order to solve the Anderson
impurity model in general multiorbital case, we use a rational interpolative
formula for the self–energy. This can be encoded into a form
–band. In
order to solve the Anderson
impurity model in general multiorbital case, we use a rational interpolative
formula for the self–energy. This can be encoded into a form
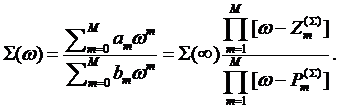 (2)
(2)
The coefficients
![]() ,
, ![]() , or, alternatively, the poles
, or, alternatively, the poles ![]() , zeroes
, zeroes ![]() and value
of the self-energy at infinity
and value
of the self-energy at infinity ![]() in this equation are to be determined. The form (2) can be also
viewed as a continuous fraction expansion but continuous fraction
representation will not be necessary for the description of the method.
in this equation are to be determined. The form (2) can be also
viewed as a continuous fraction expansion but continuous fraction
representation will not be necessary for the description of the method.
Our basic
assumption is that only a well distinct set of poles in the rational
representation (2) is necessary to reproduce an overall frequency dependence of
the self–energy. Extensive experience gained from solving Hubbard and periodic
Anderson models within DMFT at various ratios of the on–site Coulomb
interaction ![]() to the bandwidth
to the bandwidth ![]() shows the appearance of lower and upper Hubbard bands as well as
renormalized quasiparticle peak in the spectrum of one–electron excitations.
shows the appearance of lower and upper Hubbard bands as well as
renormalized quasiparticle peak in the spectrum of one–electron excitations.
It is clear
that the Hubbard bands are damped atomic excitations and to the lowest order
approximation appear as the positions of the poles of the atomic Green’s
function. In the ![]() symmetry case which is described by the Hamiltonian (1), these
energies are numerated by the number of electrons occupying impurity level,
i.e.
symmetry case which is described by the Hamiltonian (1), these
energies are numerated by the number of electrons occupying impurity level,
i.e. ![]() and the atomic Green’s function takes a simple form
and the atomic Green’s function takes a simple form
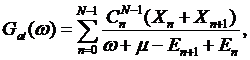 (3)
(3)
where ![]() are the
probabilities to find an atom in configuration with
are the
probabilities to find an atom in configuration with ![]() electrons
while combinatorial factor
electrons
while combinatorial factor ![]() appear due to equivalence of all states with
appear due to equivalence of all states with ![]() electrons
in
electrons
in ![]() .
.
We can represent the atomic Green’s function (3) using the rational representation (2), i.e.
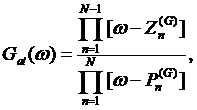 (4)
(4)
where ![]() are all
are all ![]() atomic
poles, while
atomic
poles, while ![]() denote
denote ![]() zeroes with
zeroes with ![]() being the total degeneracy. The centers of the Hubbard bands are
thus located at the atomic excitations
being the total degeneracy. The centers of the Hubbard bands are
thus located at the atomic excitations ![]() . Using
standard expression for the atomic Green’s function
. Using
standard expression for the atomic Green’s function ![]() , we
arrive to a desired representation for the atomic self–energy
, we
arrive to a desired representation for the atomic self–energy
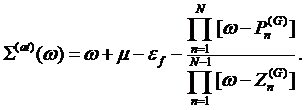 (5)
(5)
Using this
functional form for finite ![]() modifies
the positions of poles and zeroes via recalculating probabilities
modifies
the positions of poles and zeroes via recalculating probabilities ![]() which is
equivalent to the famous Hubbard I approximation.
which is
equivalent to the famous Hubbard I approximation.
We now
concentrate on the description of the quasiparticle peak which is present in
metallic state of the system. For this an extra pole and zero have to be added
in Eq (5). To see this, let us consider the Hubbard model for the ![]() case
where the local Green’s function can be written by the following Hilbert
transform
case
where the local Green’s function can be written by the following Hilbert
transform ![]() If self–energy lifetime effects are ignored, the local spectral
function becomes
If self–energy lifetime effects are ignored, the local spectral
function becomes ![]() where
where ![]() is the non–interacting density of states. The peaks of the spectral
functions thus appear as zeroes in Eq. (5) and in order to add the
quasiparticle peak, one needs to add one extra zero (denoted hereafter as
is the non–interacting density of states. The peaks of the spectral
functions thus appear as zeroes in Eq. (5) and in order to add the
quasiparticle peak, one needs to add one extra zero (denoted hereafter as ![]() to the
numerator in Eq. (5). To make the self–energy finite at
to the
numerator in Eq. (5). To make the self–energy finite at ![]() one has
to also add one more pole (denoted hereafter as
one has
to also add one more pole (denoted hereafter as ![]() ) which
should appear in the denominator or Eq. (5). Furthermore, frequently the
Hartree Fock value for the self–energy can be computed separately and it is
desirable to have a parameter in the functional form (5) which will allow us to
fix
) which
should appear in the denominator or Eq. (5). Furthermore, frequently the
Hartree Fock value for the self–energy can be computed separately and it is
desirable to have a parameter in the functional form (5) which will allow us to
fix ![]() . An obvious candidate to be changed is that self–energy pole in (5)
which is closest to
. An obvious candidate to be changed is that self–energy pole in (5)
which is closest to ![]() equal zero. Let us denote this parameter as
equal zero. Let us denote this parameter as ![]() and
rewrite the denominator of (5) as
and
rewrite the denominator of (5) as 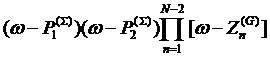 where
the product is now extended over all zeroes of the atomic Green’s functions
except the one closest to zero and two extra poles
where
the product is now extended over all zeroes of the atomic Green’s functions
except the one closest to zero and two extra poles ![]() and
and ![]() can
control the width of the quasiparticle peak and
can
control the width of the quasiparticle peak and ![]() Thus, we
arrive to the functional form for the self–energy
Thus, we
arrive to the functional form for the self–energy
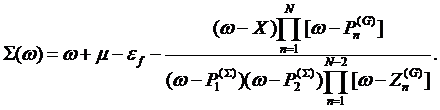 (6)
(6)
We are now
ready to list all constrains of our interpolative scheme. To fix the unknown
coefficients ![]()
![]()
![]()
![]() in Eq. (6) and to write down the linear set of equations for the coefficients
in Eq. (6) and to write down the linear set of equations for the coefficients![]() ,
, ![]() in Eq.
(2) we use the following set of conditions.
in Eq.
(2) we use the following set of conditions.
a) Hartree
Fock value ![]() In the limit
In the limit ![]() the self–energy takes its Hartree–Fock form
the self–energy takes its Hartree–Fock form
![]() (7)
(7)
Mean level
occupancy ![]() is
defined as a sum over all Matsubara frequencies for the Green’s function, i.e.
is
defined as a sum over all Matsubara frequencies for the Green’s function, i.e.
![]() (8)
(8)
where
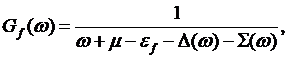 (9)
(9)
defines the
impurity Green’s function and ![]() is the
hybridization function.
is the
hybridization function.
b) Zero–frequency
value ![]() The so called Friedel sum rule establishes the relation between the
total density and the real part of the self–energy at zero frequency
The so called Friedel sum rule establishes the relation between the
total density and the real part of the self–energy at zero frequency
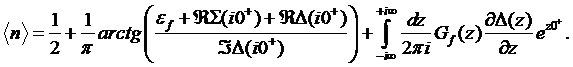 (10)
(10)
c) Quasiparticle mass renormalization value ![]() The
slope of the self–energy at zero frequency controls the quasiparticle residue,
The
slope of the self–energy at zero frequency controls the quasiparticle residue, ![]() using
the following relationship
using
the following relationship
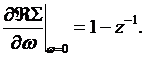 (11)
(11)
Formally, constraints (b) and (c) hold for zero temperature only but we expect no significant deviations in many regions of parameters as long as we stay at low enough temperatures. The behavior may be more elaborated in the vicinity of Mott transition.
d) Positions of Hubbard bands.
As we discussed, in order that the self–energy obeys the atomic limit and places the centers of the Hubbard bands at the positions of the atomic excitations, we demand that
![]() (12)
(12)
This condition
fixes almost all self–energy zeroes ![]() in Eq.(2)
to the poles
in Eq.(2)
to the poles ![]() However, it alone does not ensure that the weight is correctly
distributed among the Hubbard bands and that the very distant Hubbard bands
disappear. For this to occur, distant poles of Green’s function have to be
canceled out by nearby zeroes of the Green’s function. It is clear that each
pole
However, it alone does not ensure that the weight is correctly
distributed among the Hubbard bands and that the very distant Hubbard bands
disappear. For this to occur, distant poles of Green’s function have to be
canceled out by nearby zeroes of the Green’s function. It is clear that each
pole ![]() far from the Fermi level has to be accompanied by a nearby zero
far from the Fermi level has to be accompanied by a nearby zero ![]() in order
the weight of the pole be small. Thus, the self–energy has poles at the
position of Green’s function zeroes which can be encoded into the constrain
in order
the weight of the pole be small. Thus, the self–energy has poles at the
position of Green’s function zeroes which can be encoded into the constrain
![]() (13)
(13)
We want to
keep this property of the self–energy for finite ![]() and thus
demand that self–energy diverges (when lifetime effects are kept, it only
reaches a local maximum) at the zeros of the functional form (3) of
and thus
demand that self–energy diverges (when lifetime effects are kept, it only
reaches a local maximum) at the zeros of the functional form (3) of ![]() Note
that the relationship (13) holds (approximately) for frequency
Note
that the relationship (13) holds (approximately) for frequency ![]() larger
than the renormalized bandwidth
larger
than the renormalized bandwidth ![]() Therefore
the information about one
Therefore
the information about one ![]() which lies close to
which lies close to ![]() is
omitted and replaced by the information about
is
omitted and replaced by the information about ![]()
![]() and
and ![]() as it is
done by separating
as it is
done by separating ![]() and
and ![]() in the denominator of Eq. (6).
in the denominator of Eq. (6).
We can now
write down a set of linear equations for all unknown coefficients in the
expression (2). There is total ![]() of
parameters
of
parameters ![]() and
and ![]() where we can always set
where we can always set ![]() The
conditions a),b),c) give
The
conditions a),b),c) give
![]() (14)
(14)
![]() (15)
(15)
![]() (16)
(16)
![]() (17)
(17)
According to
condition d) we can use all ![]() poles
poles ![]() and
and ![]() zeroes
zeroes ![]() The zero
The zero
![]() closest to
closest to ![]() is dropped out. This brings additional
is dropped out. This brings additional ![]() equations
for the coefficients and makes
equations
for the coefficients and makes ![]() as the degree of the rational interpolation which are written below
as the degree of the rational interpolation which are written below
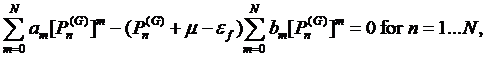 (18)
(18)
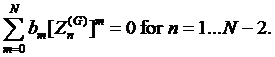 (19)
(19)
Note that
while ![]() may be rather large, the actual number of poles contributing to the
self–energy behavior is indeed very small. We can directly see this from Eq.
(5) which uses all
may be rather large, the actual number of poles contributing to the
self–energy behavior is indeed very small. We can directly see this from Eq.
(5) which uses all ![]() poles
poles ![]() fulfilling
Eq. (12) and uses
fulfilling
Eq. (12) and uses ![]() zeroes
zeroes ![]() directly
related to
directly
related to ![]() poles
poles ![]() Clearly, when the
spectral weight of the atomic excitation becomes small, the corresponding
Clearly, when the
spectral weight of the atomic excitation becomes small, the corresponding ![]() becomes
close to
becomes
close to ![]() and the cancellation occurs. Therefore in realistic situations when
only the upper and lower Hubbard bands have significant spectral weight along
with the quasiparticle peak, the actual degree of the polynomial expansion is
either two or three. However, it is advantageous numerically and cheap
computationally to keep all poles and zeroes in Eq. (6) because the formula
automatically distributes spectral weight over all existing Hubbard bands.
and the cancellation occurs. Therefore in realistic situations when
only the upper and lower Hubbard bands have significant spectral weight along
with the quasiparticle peak, the actual degree of the polynomial expansion is
either two or three. However, it is advantageous numerically and cheap
computationally to keep all poles and zeroes in Eq. (6) because the formula
automatically distributes spectral weight over all existing Hubbard bands.
In the limit
when ![]() the self–energy automatically translates to the non–interacting
one. The atomic poles get close to each other but, most importantly, their
spectral weight goes rapidly to zero as it gets accumulated within the
quasiparticle band.
the self–energy automatically translates to the non–interacting
one. The atomic poles get close to each other but, most importantly, their
spectral weight goes rapidly to zero as it gets accumulated within the
quasiparticle band.
In the Mott
insulating regime, the conditions b)
and c) drop out while all poles ![]() and zeroes
and zeroes ![]() can be used to
determine the interpolation. However, in this regime it does not matter whether
one of
can be used to
determine the interpolation. However, in this regime it does not matter whether
one of ![]() closest to
closest to ![]() is dropped out or
kept, since we can always replace this information by information about
is dropped out or
kept, since we can always replace this information by information about ![]() Therefore
the Mott transition can be studied without changing the constraints.
Therefore
the Mott transition can be studied without changing the constraints.
We thus see
that in the insulating case the self–energy correctly reproduces the well–known
result of the Hubbard I method where the Green’s function is computed after Eq.
(9) with atomic self–energy. If the lifetime effects are computed, the
parameters ![]() and
and ![]() become complex and the
Hubbard bands will acquire an additional bandwidth. This effect is evident from
the simulations using various perturbative or QMC impurity solvers and can be
in principle incorporated into the interpolative formulas (2) or (6). However,
in in the current implementation we omit it for simplicity.
become complex and the
Hubbard bands will acquire an additional bandwidth. This effect is evident from
the simulations using various perturbative or QMC impurity solvers and can be
in principle incorporated into the interpolative formulas (2) or (6). However,
in in the current implementation we omit it for simplicity.
We thus see
that the interpolational scheme is defined completely once a prescription for
obtaining parameters such as ![]()
![]() as well as poles
as well as poles ![]() ,
and zeroes
,
and zeroes ![]() is given. For this purpose we will test two commonly used methods:
SBMF method due to Gutzwiller [1] as described by Kotliar and Ruckenstein [2]
and the well–known Hubbard I approximation [3]. We compare these results
against more accurate but computationally demanding quantum Monte Carlo calculations and establish the
procedure to extract all necessary parameters.
is given. For this purpose we will test two commonly used methods:
SBMF method due to Gutzwiller [1] as described by Kotliar and Ruckenstein [2]
and the well–known Hubbard I approximation [3]. We compare these results
against more accurate but computationally demanding quantum Monte Carlo calculations and establish the
procedure to extract all necessary parameters.
Note that once
the constraints such as ![]() are computed
from a given approximate method, some of the quantities such as the total
number of particles,
are computed
from a given approximate method, some of the quantities such as the total
number of particles, ![]() , and the value of the
self–energy at zero frequency,
, and the value of the
self–energy at zero frequency, ![]() can be computed fully
self–consistently. They can be compared with their non–self–consistent values. If
the approximate scheme already provides a good approximation for
can be computed fully
self–consistently. They can be compared with their non–self–consistent values. If
the approximate scheme already provides a good approximation for ![]() and
satisfies the Friedel sum rule, the self–consistency can be avoided hence
accelerating the calculation. Indeed we found that inclusion of the
self–consistency improves the results only marginally except when we are in a
close vicinity to the Mott transition but here we do not expect that our simple
interpolative algorithm is very accurate.
and
satisfies the Friedel sum rule, the self–consistency can be avoided hence
accelerating the calculation. Indeed we found that inclusion of the
self–consistency improves the results only marginally except when we are in a
close vicinity to the Mott transition but here we do not expect that our simple
interpolative algorithm is very accurate.
[1] M. Gutzwiller, Phys. Rev. 134, A923 (1964).
[2] G. Kotliar and A. E. Ruckenstein, Phys. Rev. Lett. 57, 1362 (1986).
[3] J. Hubbard, Proc. Roy. Soc. (London) A281, 401 (1964).
Running program
· Compile program typing "make" at the source files location. Makefile was tested on Linux OS using PGI compiler. Please adjust it depending on operating system and compiler used.
· Edit “input" file which has the following structure:
|
1 |
IMOD |
|
0.0 |
EF |
|
2.0 |
U |
|
0.016 |
TEMP |
|
4 |
NDEG |
|
500 |
NMSB |
|
500 |
OMAX |
|
500 |
NOMG |
|
50 |
WEND |
|
F |
COMPUTE_REAL |
where IMOD = 1 is the Hubbard Model, IMOD=3 the Anderson impurity model, EF is the impurity level, U is value of Coulomb repulsion, TEMP is temperature to be used used to create for Matsubara points grid, NDEG is the degeneracy, NMSB is the number of Matsubara point, OMAX is the imaginary frequency cutoff, NOMG is the number is real frequency points, WEND is the real frequency cutoff, COMPUTE_REAL is flag once is “True" tell program to produce the self-consistent solution (last iteration) on real axis.
1. Program’s input consists from one more file (provided the Anderson impurity model item is chosen in “input" file):
“delta.dat" containing the hybridization function.
The structure is the following:
|
Column # |
Value |
|
1 |
Frequency |
|
2 |
Re Delta |
|
3 |
Im Delta |
2. Run the program executable (“main" is the default name).
3. Program’s output consists from the following files:
1) “gfsig_iw.dat" containing Green’s function (GF) and the self-energy (SE) on Matsubara axis
2) “gfsig_re.dat" containing Green’s function (GF) and the self-energy (SE) on real axis provided flag "COMPUTE_REAL" is “TRUE".
They have the same structure:
|
Column # |
Value |
|
1 |
Frequency |
|
2 |
Re GF |
|
3 |
Im GF |
|
4 |
Re SE |
|
5 |
Im SE |
3) “grid_re.dat" containing real frequency grid for the hybridization function (delta).
4) “grid_im.dat" containing Matsubara frequency grid for the hybridization function (delta).
Both files have the same structure:
|
Column # |
Value |
|
1 |
Frequency |
Program content
|
Makefile |
main make file |
|
dmf_cmpdiag.f |
The solution of the generalized eigenvalue problem |
|
imp_sunatm.f |
Solves Anderson impurity model, returns GF and SE. |
|
imp_sunmas.f |
Computes effective mass renormalization |
|
imp_sunrat.f |
Finds rational interpolation or self-energy |
|
lib_broy6.f |
Broyden mixing. |
|
lib_cinv.f |
Finds inverse of square matrix. |
|
lib_csplines.f |
Splines complex function from one to another grid. |
|
lib_deriv1.f |
Calculates radial derivative. |
|
lib_morefun.f |
Calculations of a few auxiliary functions |
|
lib_pade.f |
Realization of Pбde procedure. |
|
lib_pcoefs.f |
Finds poles |
|
lib_pzeros.f |
Finds zeros |
|
lib_simq.f |
Finds solution of a set of linear equations |
|
lib_splin3.f |
Computed a natural spline approximation of third order. |
|
mod_common.f |
File containing common modules used across the program. |
|
mod_dimart.f |
File containing common modules used across the program. |
|
mod_init.f |
File containing common modules used across the program. |
|
sun.f |
Main program. |