Extracting LDA band structure parameters
To calculate transport parameters, including the optical conductivity as well as to calculate the photoemission spectra one needs make momentum summation of Green’s function (GF) or their convolutions. To do so one needs to make k-summation of DOS or the local Green’s function of a type:
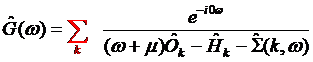
where ![]() is the chemical potential,
is the chemical potential, ![]() is the overlap matrix,
is the overlap matrix, ![]() is the Hamiltonian and
the
is the Hamiltonian and
the ![]() is the self-energy
matrix. In the LDA calculation the self-energy is represented by the
Hartree-Fock constant while in the DMFT calculations it is frequency dependent function
is the self-energy
matrix. In the LDA calculation the self-energy is represented by the
Hartree-Fock constant while in the DMFT calculations it is frequency dependent function![]() . Surely this
integration/summation is the part of the LDA program we described in the
previous section but very often it is quite convenient to have a separate
routine/driver which does it outside one big program as it is much easier to
change it in accord to a specific task.
. Surely this
integration/summation is the part of the LDA program we described in the
previous section but very often it is quite convenient to have a separate
routine/driver which does it outside one big program as it is much easier to
change it in accord to a specific task.
But to make the summation we still use some information from LDA program i.e. the Hamiltonian, the overlap matrix, information about symmetry of a studied material. Program which prints all the necessary files for k-summation driver is the same as we described in the previous section with the only difference in printouts included. It means that one can make the self-consistent LDA calculation whether using this program or the previous one. And then one needs to make optical calculations to extract all the parameters. Run file should look something like this:
/home/user/LMTO/main <<!
latio3
ini+str+scs
scf+out
!
mv latio3.scf latio3.scs
mv latio3.out latio3.out_scf
/home/user/LMTO/main <<!
latio3
ini+str+scs
opt+out
!
mv latio3.out latio3.out_opt
(change /home/user/LMTO/ to the appropriate path to the
program and latio3 to
the studied material)
Once the program run one should get the following files as an output:
|
1. ham.dat |
The Hamiltonian |
|
2. olp.dat |
The overlap |
|
3. bz.dat |
Brillouin zone data |
|
4. grp.dat |
Space group data |
|
5. wig.dat |
Space group data |
|
6. idx.dat |
Auxiliary index pointer |
|
7. opar.dat |
|
|
8. Sk.dat |
Structure constants |
|
9. vel.dat |
Velocity matrix |
1. Hamiltonian
data (ham.dat).
This file contains the Hamiltonian matrix at each irreducible mesh
point in the Brillouin zone. To facilitate and standardize the rotations of the
Hamiltonian within the Brillouin zone the Hamiltonian is always assumed to be
written in a spherical harmonics basis.
|
Quantity |
Variable |
|
Number of irreducible k-points |
nkp |
|
Dimension of
Hamiltonian |
ndim |
|
Hamiltonian |
hamf (1:ndim,1:ndim,1:nkp) |
Table I:
Parameters available from ham.dat file.
Default energy unit on the Hamiltonian is Rydberg. Ndim=2 norb is
the spin-orbital case is considered.
2. Overlap
matrix data (olp.dat).
This file contains the overlap matrix at each irreducible mesh point
in the Brillouin zone. The dimension of this matrix should be equal to the
dimension of the Hamiltonian and the number of irreducible k-points should also
match with the Hamiltonian data.
|
Quantity |
Variable |
|
Number of irreducible k-points |
nkp |
|
Dimension of overlap
matrix |
ndim |
|
Overlap matrix |
olap (1:ndim,1:ndim,1:nkp) |
Table II: Parameters
available from olp.dat file.
3. Brillouin zone data
(bz.dat).
The file bz.dat contains
the data on the Brillouin zone. It contains such information as the lattice
constant, the mesh divisions, the irreducible k-points and tetrahedra as well as information about the group
transformations that connect each k-point
to its irreducible representative.
|
Quantity |
Variable |
|
Lattice constant |
latcon |
|
Number of irreducible k-points |
nkp |
|
Number of irreducible
tetrahedra |
ntet |
|
Total number of k-points |
nktot |
|
Number of k-points along each direction |
ndiv (1:3) |
|
Coordinate array for irreducible k-points |
bk (1:3,1:nkp) |
|
Integration weight array for irreducible
k-points |
wk (1:nkp) |
|
Pointer from each irreducible tetrahedron
to its multiplicity, irreducible k-point
corners and type |
itt (1:6,1:ntet) |
|
kcut matrices for all tetrahedron types
(see Figure) |
kcut (1:3,1:4,1:6) |
|
Pointer from irreducible k-point to the four nearest
irreducible neighbors in each direction |
nbr (1:3,1:4,1:nkp) |
|
Pointer from irreducible k-point to the group operation that
rotates the irreducible neighbor to the real neighbor |
grop (1:3,1:4,1:nkp) |
|
Bravais lattice vectors, multiplied by
the division along the reciprocal direction in Brillouin zone |
rvec (1:3,1:3) |
|
Brillouin zone
minicell vectors (i.e. |
mvec (1:3,1:3) |
|
Pointer from each mesh points to its
irreducible representative |
kirrptr (0:ndiv(1)-1,0:ndiv(2)-1, 0:ndiv(3)-1) |
|
Pointer from each mesh point to the group
operation that takes the irreducible representative to that point |
gropptr (0:ndiv(1)-1,0:ndiv(2)-1, 0:ndiv(3)-1) |
Table III.
Parameters available from bz.dat file.
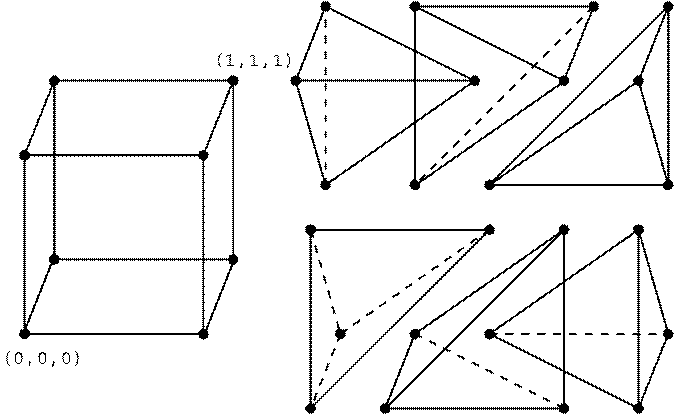
Figure I: One of four different ways of splitting the unit cube
into six tetrahedral. The common diagonal is chosen from ![]() to
to ![]() . The coordinates of a given corner
. The coordinates of a given corner ![]() of each tetrahedron
of each tetrahedron ![]() are given by the pointer kcut(m,
are given by the pointer kcut(m,![]() ,
,![]() )
)![]() where
where ![]() .
.
4. Group
operations (grp.dat).
The file grp.dat contains
information about the space group operations of the crystal. Here we read in
both the number of atoms and the number of group operations that exist. In
principle the maximum number of symmetry operations is 48 (cubic) but in low
symmetry crystals they could be less and if one is using a reducible (i.e. not
minimal) unit cell they could be more. To each of the symmetry operation
corresponds a group operation element but since the maximum number of group
elements is 48, two or more symmetry operations could correspond to a given
group element. In addition to knowing the correspondence between the symmetry
operations and the group elements one must know how the atoms move under the
action of the symmetry operation if there are more than one atom of a given
type in the unit cell. There is also the possibility that the symmetry
operation takes the atom out of the unit cell and then it must be translated
back by a lattice vector to the original unit cell. Since the Hamiltonian is in
the Brillouin zone these translations must be known because they introduce
phase factors into the equation for the transformation of the Hamiltonian.
|
Quantity |
Variable |
|
Number of symmetry
operations |
ngroup |
|
Number of atoms in unit cell |
natom |
|
Symmetry operation to group element
pointer |
ikov (1:ngroup) |
|
Atom to atom mapping for each symmetry
operation |
igatom (1:natom,1:ngroup) |
|
Back to cell translation vectors for each
atom and symmetry operation |
rdc (1:3,1:natom,1:ngroup) |
Table IV. Parameters available from grp.dat file.
5. Group
operations (wig.dat).
The file wig.dat contains
Wigner matrices for spherical harmonics angular momenta up to![]() . This is needed to both rotate the
Hamiltonian and the overlap matrix and to create the vector rotation matrices
for each group operation. It contains \the number of group operations and the
maximum angular momentum (should be always 3). The Wigner matrix is stored as a
vector in the computer and the mapping from the angular momentum labels,
. This is needed to both rotate the
Hamiltonian and the overlap matrix and to create the vector rotation matrices
for each group operation. It contains \the number of group operations and the
maximum angular momentum (should be always 3). The Wigner matrix is stored as a
vector in the computer and the mapping from the angular momentum labels,![]() , to the vector component,
, to the vector component, ![]() , is given by:
, is given by: ![]()
|
Quantity |
Variable |
|
Number of group
operations |
ng |
|
Maximum angular
momentum |
lmax |
|
Wigner matrices |
wigmat (1:imax,1:48) |
Table V. Parameters available from wig.dat file.
|
|
|
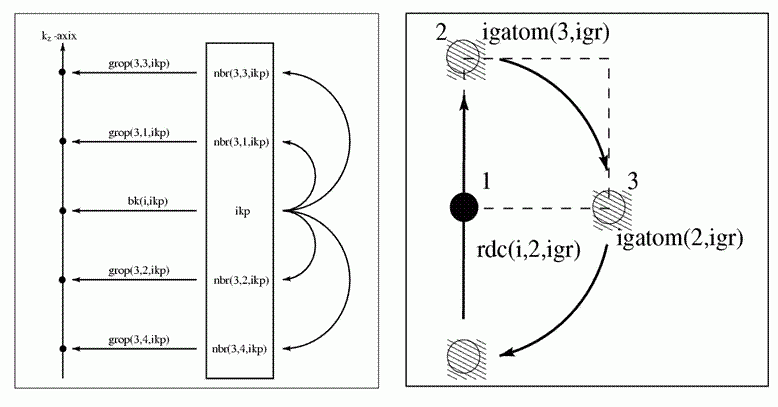
Figure II:
Left figure: An illustration of the
information encoded in the arrays nbr and grop along the 3rd (![]() ) direction. Right figure: An
illustration of the back-to-cell vectors rdc that take an atom that is rotated
by the transformation igr out of the unit cell back to its equivalent position
in the unit cell.
) direction. Right figure: An
illustration of the back-to-cell vectors rdc that take an atom that is rotated
by the transformation igr out of the unit cell back to its equivalent position
in the unit cell.
In addition to the parameters in Table V available from the file bz.dat there are two more parameters that
are initialized using information in bz.dat. One of them is the total number of
tetrahedra which is computed from the variable itt but should of course
be equal to![]() . The other one is a pointer from
each corner of every tetrahedron to its irreducible representative.
. The other one is a pointer from
each corner of every tetrahedron to its irreducible representative.
|
Quantity |
Variable |
|
Total number of
tetrahedra |
mtet |
|
Pointer from each tetrahedron corner to its
irreducible representative |
coridx (1:4,1:6,1:ndiv(1),1:ndiv(2),1:ndiv(3)) |
Table VI: Additional Parameters
available initialized from data in bz.dat.
6. Auxiliary index data (idx.dat).
The file idx.dat contains pointers from the computer representation
of the physical quantities (Hamiltonian etc.) as array to the corresponding
atoms and angular momenta. Here we also get information about how many sorts of
atoms there are, a pointer from each atom to its sort and what is the maximum
angular momentum for a given atomic sort. We also have a logical array that
indicates if some angular momenta are omitted from the Hamiltonian. This is
useful if we want to consider simplified models where certain angular momenta
have been ignored, usually because they are of much to low energy to be of
interest.
|
Quantity |
Variable |
|
Number of atoms in unit cell |
natom |
|
Number of atomic sorts |
nsort |
|
Pointer from each atom to its sort |
is (1:natom) |
|
Pointer from each sort to its maximum
angular momentum |
ilsb (1:nsort) |
|
Logical array indicating whether a given
angular momentum is present for a given sort of atoms |
ltpan (1:lmax,1:natom) |
|
A pointer from each orbital state of
every to its position in the Hamiltonian matrix |
lmtind (1:(lmax+1) |
Table VII:
Parameters available from idx.dat file.
7. Factorization of the
overlap (opar.dat):
Files opar.dat and Sk.dat are used to orthonormalize
(almost) the overlap matrix.
|
Quantity |
Variable |
|
Maximum angular momentum used |
l_max |
|
Number of atomic sorts |
nsort |
|
Number of spins |
nspin (=2 for
spin-orbit) |
|
Transformation matrixes |
moo(4,0:l_max,nsort,nspin) |
Table VIII. Parameters available
from opar.dat file.
8. Structure constants (Sk.dat):
The file Sk.dat contains the structure constants data.
|
Quantity |
Variable |
|
Maximum angular momentum used +1 squared |
(l_max+1) |
|
Number of atoms in unit cell |
natom |
|
Lattice constant |
latcon |
|
Structure constants |
Stk(1:ndim,1:ndim,1:nkp) |
Table VIII. Parameters available from
Sk.dat file.
9. Velocity
matrix (vel.dat).:
The vel.dat is the velocity matrix. It is not used in calculation of the k-integral. This information is used in the optical calculations.