Calculation of
DOS and Green’s function*
The definitions of the one-electron
Green’s function for interacting system is
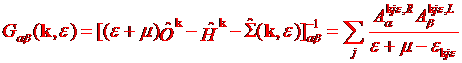 (1.1)
(1.1)
where
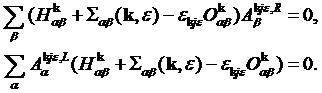
Here “L” denotes left eigenvector ![]() to distinguish it from the right “R” eigenvector
to distinguish it from the right “R” eigenvector![]() ,
,
![]() is the chemical potential,
is the chemical potential, ![]() are the eigenvalues,
are the eigenvalues, ![]() is the self-energy,
is the self-energy, ![]() is the Hamiltonian
is the Hamiltonian
and ![]() is the overlap matrix
which takes into account the non-orthogonality of the basis set
is the overlap matrix
which takes into account the non-orthogonality of the basis set
![]() ,
,
and is defined as follows
![]() .
.
The orthonormality condition is
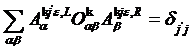 ,
,
or in the matrix form
![]() .
.
Now one can easy see by inspection
that the Hamiltonian can be written in the form:
![]() ,
,
where ![]() is a diagonal matrix of eigenvalues. Using the above expression for the Hamiltonian
one can easily prove the above equation for the Green’s function:
is a diagonal matrix of eigenvalues. Using the above expression for the Hamiltonian
one can easily prove the above equation for the Green’s function:
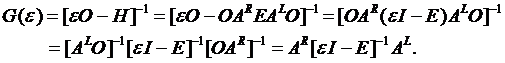
In direct space, formula for the Green’s
function is
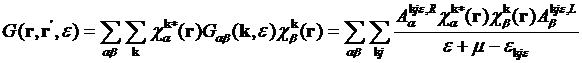 ,
,
or
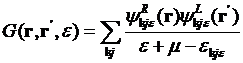 ,
,
where the wave functions are
![]() ,
,
which also satisfy to the following equations
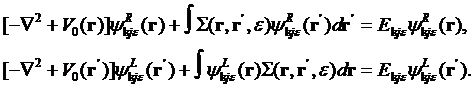
The Equation for the GF in direct space is
just the Fouriesr transformation of ![]() taking into account
the basis set equation:
taking into account
the basis set equation:
![]() .
.
Density can be found from the Green’s
function directly
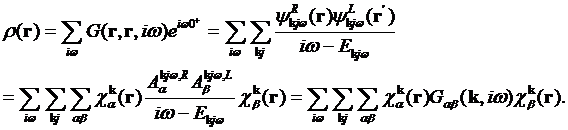
Integration over the space gives the interacting density matrix which should be
defined with the overlap matrix
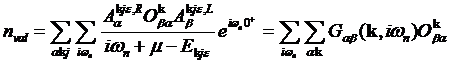 ,
,
in order to preserve the sum rule
related to the total number of electrons
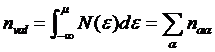 .
.
Note, that as soon as we assume that
our basis set is orthonormal, we restore ordinary definitions for all the
relevant quantities discussed above.
The total density of states (TOS) is defined as
![]() .
.
Unfortunately, the definition
![]() ,
,
has a problem since it does not have
proper symmetry transformation. By this we mean that
![]() ,
,
where ![]() are Wigner matrices
and
are Wigner matrices
and![]() is
any group operation. Assuming by
is
any group operation. Assuming by ![]() the rotation of the entire Brillouin zone
the rotation of the entire Brillouin zone
![]() ,
,
we see that we cannot prove this equality.
We can easily prove this for objects like
![]()
A possible way to resolve it can be found
if the overlap matrix can be represented as
![]() (1.2)
(1.2)
where ![]() is the structure
constants and
is the structure
constants and ![]() are the overlap
matices of Hankel and Bessel functions
are the overlap
matices of Hankel and Bessel functions ![]() .
.
This representation is true in the Atomic
Sphere Approximation (ASA) only. If such factorization possible, we can define
the non-interacting density of states as
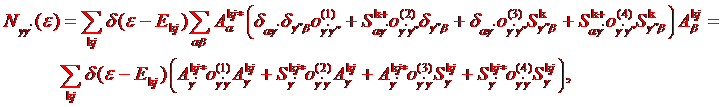 (1.3)
(1.3)
where by ![]() we denoted
convolutions
we denoted
convolutions
![]() .
.
It is clear
that each term obeys the required transformation property, and so does the
resulting ![]()
Next we consider the basis
orthonormalization procedure. One can show that the overlap matrix can be
presented in the form
![]() (1.4)
(1.4)
where we
made the following definitions
![]() .
.
Here h and j are matrices which are
calculated by minimization such that Eq. (1.4) is best satisfied.
Let us
denote transformation ![]() which we use to bring
the system to the orthonormal basis
which we use to bring
the system to the orthonormal basis
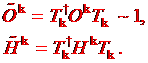 (1.5)
(1.5)
In the almost
orthonormal basis the Green’s function reads as
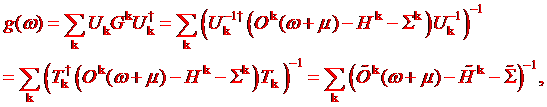 (1.6)
(1.6)
where
![]() (1.7)
(1.7)
Having GF g we can compute the density of states as
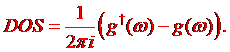 (1.8)
(1.8)
Using GF and the self-energy
we can also compute the Weis GF using the following relation:
![]() . (1.9)
. (1.9)
The impurity levels can be obtained from the frequency expansion of the
impurity and local GFs:
![]() , (1.10)
, (1.10)
where matrix
contains![]() is the Hartree-Fock values of the self-energy,
is the Hartree-Fock values of the self-energy, ![]() is the double counting term. As we see to
compute the impurity levels we need to know the following averages (which are
matrixes):
is the double counting term. As we see to
compute the impurity levels we need to know the following averages (which are
matrixes): ![]() .
.
If we are interested only in diagonal elements of the impurity matrix then the
expression (1.10) is simplified to
![]() , (1.11)
, (1.11)
where values of ![]() are provided by the
k-sum driver/program.
are provided by the
k-sum driver/program.
* Most of the equations
in the text related to LMTO ASA method can be found in
O. K. Andersen, Linear methods in band theory, Phys. Rev. B 12, 3060 (1975).
Last updated @ Sept 6 2004