Next: About this document ...
Up: NCA_descrip
Previous: How to generate the
The program can be compiled on linux PC by typing "make
PLATFORM=PC RELEASE=1". The second option produces the optimized code.
A short help can always be obtained by executing the program and
giving no arguments. Thus typing nca gives the following
output
|
Sig = sig.inp |
# The name of the input Self-energies |
|
Ac = Ac.inp |
# The name of the input bath function |
|
cix = cix.dat |
# The name of the input file containing information about bands and their degeneracy |
|
out = . |
# The name of the output directory |
|
gloc = gloc.out |
# The name of the output Green's function |
|
sig = sig.out |
# The name of the output Self-energy |
|
Ed = -2 |
# Energy level |
|
U = 4 |
# Coulomb repulsion |
|
T = 0.2 |
# Temperature |
|
Q = 1 |
# Default average Q in grand-canonical ansamble |
|
alpha = 0.5 |
# The fraction of the new self energy to be used in the next iteration |
|
max_diff = 1e-06 |
# Criterium to finish the procedure |
|
max_steps = 300 |
# Criterium to finish the procedure |
|
StartLambda = -1 |
# Where to start searching for the lambda0 |
|
EndLambda = 1 |
# Where to stop searching for the lambda0 |
|
dLambda = 0.1 |
# Step in searching for the lambda |
|
followPeak = -3 |
# Wheather to determin zero frequency from the diverging pseudo-particle |
|
|
(-2: lamdba0==StartLambda, -1: Q==Q0, 0: follow b, 1: follow f, 2: foolow a) |
|
prt = 1 |
# Weather to print intermediate results |
|
MissDopSt = -100 |
# Missing doping due to projection and finite mesh starting at MissDopSt |
All control parameters are printed with there default values. After
the symbol # a short description of each parameter is also
given. Below we will give some more description to each parameter but
let us run an example program first.
The necessary files can be found in the subdirectory PC/work
and PC/work/start. The example is for the f-electrons with
around 6 electrons on the impurity. The crystal field splits the 7
band problem into 3+3+1 band problem (To generate the input file one
thus needs to use 3 bands and degeneracy 6,6,2). All the impurity
states with 4,5,6 and 7 electrons on the impurity are keept
which results in a 64 nonequivalent pseudoparticle (atomic)
states. The necessary coefficients 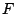 's to run the program are
already prepared in the file PC/work/start/cix4567.cix.
's to run the program are
already prepared in the file PC/work/start/cix4567.cix.
To run the program, one also needs a bath spectral function (it is in
PC/work/start/Ac.12) and starting guess for the pseudo
self-energies (can be found in PC/work/start/Sigma.000).
The input bath spectral function contain four columns, frequency and
three nonequivalent bath spectral functions. The pseudo self-energy
files contains 65 columns, in addition to frequency, a guess
self-energy for all 64 pseudo particles.
An example of how to run the program can be found in the file
history.nca in a subdirectory PC/work and reads
../nca Sig=start/Sigma.000 Ac=start/Ac.12 cix=start/cix4567.cix U=4 T=0.1 "Ed={-20.67,-20.58,-20.64}"
Each time the program is executed, the command line is saved
(appended) to the file history.nca for easier restart of a
job at a later time.
In the example, the self-consistency is reached after about 30
iterations (the difference between the steps below 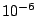 ) and the
following results are ready to be plotted
) and the
following results are ready to be plotted
- Impurity Green's function is in the file gloc.out (the
filename can be changed in the command line)
- The physical self-energy is in the file sig.out (the filename
can also be chnaged if desired)
- the latest pseudo self-energies are in the file Sigma.000
- the latest pseudo spectral functions are in the file Spec.000
- pseudo self energies for each iteration are saved in a file Sigma.xxx
- pseudo spectral functions for each iteration are saved in a file Spec.xxx
The spectral function obtained in this example is plotted in figure 1.
At each iteration, the following information is written in the
standard output
Figure 1:
The spectral function in the example.
![\includegraphics[width=0.50\linewidth angle=0]{spectra1.eps}](img74.png) |
- The value of
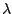 discussed in the chapter 1. (lambda=-64.8101064793649)
discussed in the chapter 1. (lambda=-64.8101064793649)
- The value of
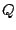 that was used (Q=9438 which is total number
of states here)
that was used (Q=9438 which is total number
of states here)
- The occupancy of each atomic state (the sum of all numbers is
unity)
- Total impurity occupancy (nd: 5.73398078352997)
- Norm of every pseudo spectral function (should be close to
one if mesh resolves the structure and was chosen
correctly).
- Impurity occupancy per each band (0: 2.5582 1: 2.4337 2:
0.7420)
- The difference between last two steps (Difference between steps: 6.768e-07).
Finally, let me give some more details of every parameter of the
program
- Sig: The filename of the input guess for the pseudo self-energies
- Ac: The filename of the input bath spectral function
- cix: The input file that was generated by the
program generate and discussed in chapter 2.
Note that the number of columns in the Sig file should match the
number of pseudoparticle states from cix-file and the number of
columns in Ac file should match the number of baths in the cix-file.
- out: The name of the output directory. Default is the current
directory.
- gloc : Filename of the output local Green's function
- sig : Filename of the output local self-energy
- Ed : Impurity energies. The number of impurity energies
should match the number of baths (one impurity energy for each
nonequivalent bath). If not, the program assumes that the rest of
the energies are equal to the last specified energy.
They have to be given in curly brackets and in the shell, additional
quotation marks should be used, for example "Ed={-3,-3,-3}".
- U : The Coulomb repulsion
- T : Temperature
- Q : The desired charge explained in chapter
1. This is used only when followPeak is set to -1.
- alpha : The mixing parameter for the pseudo self-energy
- max_diff : The difference between successive self-energies has
to be smaller than this number to stop iterating.
- max_steps : The number of iterations is always less than this
number (regardless of max_diff) to avoid infinite loops.
- StartLambda : If is fixed number, one needs to find the
corresponding as explained in chapter 1. For
that, one needs to solve a nonlinear equation and the solution is
always looked for in the interval StartLambda and
EndLambda. StartLambda should be enough negative to catch the
solution. If the solution is not found, tray to decrease this number
(more negative) and if the solution is found, keep the new
value. However, very small StartLambda can result in worse
performance.
- EndLambda: Explained above
- dLambda : If one looks for the allover the interval
[StartLambda,EndLambda], the solution is very unlikely to be
cached. The reason is that when a guess is larger than the
that corresponds to the solution, the spectral functions
are more diverging and therefore the spectral functions are much worse
resolved. The strategy is to start looking for solution at most
negative lambda (StartLambda) and slowly move up until the solution
is bracket and then use the Newton procedure. The small step when
bracketing the solution is dLambda. Can be much decreased if the
solution is hard to catch (very close to the Mott transition).
- followPeak: There are four strategies implemented to fix zero
frequency of the pseudoparticle quantities. When followPeak is -3,
the is fixed at the total number of all local states and
is then looked for. If the followPeak is -2, is
chosen to be equal to
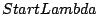 and is calculated from the
equation (16). If followPeak is -1, is fixed to the input
(specified in the command line, the parameter Q above) and the
corresponding is looked for. If follow peak is a
non-negative number
and is calculated from the
equation (16). If followPeak is -1, is fixed to the input
(specified in the command line, the parameter Q above) and the
corresponding is looked for. If follow peak is a
non-negative number 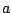 , the zero frequency (and ) is
chosen such that the maximum of the pseudoparticle spectral
function with index is at zero frequency.
, the zero frequency (and ) is
chosen such that the maximum of the pseudoparticle spectral
function with index is at zero frequency.
- prt : If prt is 1, pseudo self-energy and spectral functions
are printed at each iteration. It can also be set to 0.
- MissDopSt: Sometimes the mesh on which the electron spectral
function is calculated, does not catch all the spectral weight at
negative frequencies, or alternatively, there is some weight of
spectral function lost due to projection (if we take occupancies
n-1,n,n+1,... and n-2 was still having some nonzero probability). In
this case, we want to calculate how much weight is missing when
integrating spectral function on the output mesh. This is very
important in the real material calculation, because the missing
weight should be added to the total number of electrons below the
Fermi level, to get the right occupancy and right chemical potential.
The parameter should be set to the point below which the weight is
considered to be lost.
Next: About this document ...
Up: NCA_descrip
Previous: How to generate the
Kristjan Haule
2004-08-23